International Classification of Rhabdomyosarcoma
Superior prognosis: Botryoid Rhabdomyosarcoma
Spindle cell Rhabdomyosarcoma
Intermediate prognosis: Embryonal Rhabdomyosarcoma
Poor prognosis: Alveolar Rhabdomyosarcoma
Undifferentiated sarcoma

Introduction: Pigmented neuroepithelial tumor is a rare tumor. About 200 cases has been reported till now. It is characteristically occur in maxilla & has a typical bluish color due to the presence of melanin. It is benign tumor with 2% chances of malignancy. It is locally aggressive. Krompecker described it first in 1918 Many names given as the cell of origin was not clear ( Pigmented ameloblastoma, Retinal anlage tumor, Melanotic adamantinoma, Retinal choristoma)
Presentation: Majority present in1st year of life. Median age of occurrence is 4.3 months. There is no sexual predilection.
• Swelling in the region of oral cavity
• Often feeding & sucking impaired
Investigations: There are no characteristic radiological findings so pathology remains the cornerstone of diagnosis.
Plain radiograph & CT/MRI: Well-circumscribed, low-density lesion without calcification typically arising from maxilla. As the tumor grows, bone is destroyed suggesting a malignant process.
Treatment: Treatment of choice is complete surgical excision. Developing teeth & surrounding tissues may need to be sacrificed to attain an adequate surgical margin.
Recurrence rate is 10-60%.Radiotherapy & Chemotherapy are used for inoperable recurrence or margin positive resection. Close follow up is necessary to detect recurrence
Permanent reconstruction can be done after growth is completed
Introduction: This is the commonest cancer in children. Types of leukemia seen in pediatric age group are
1. Acute lymphoblastic leukemia
2. Acute non-lymphoblastic leukemia
3. Chronic myeloid leukemia
Acute lymphoblastic leukemia:
Incidence: ALL is the most common type of leukemia in children.The incidence of ALL is about 30 cases per million people per year. The peak age of developing ALL is 2-6 years.
Etiology:
1. Genetic factor- It is seen in identical twins, Fanconi's anamia, Down's & Klinefelter's syndrome,
2. Immunologic factor- Ataxia telangiectasia
3. Environmental factor-Exposure to radiation, chemotherapeutic agents like alkalyting agents.
4. Viral infection- Epstein-Barr virus
Clinical features:
1. Anemia (fatigue, irritability, pallor)
2. Thrombocytopenia (bleeding, petechiae)
3. Neutropenia (fever)
4. Lymphadenopathy
5. Hepatospenomegaly
6. Joint pains
7. Headache, vomitings (Brain involvement)
Diagnosis:
1, Complete blood count-Eleveted leucocyte counts
2. Serum levels of uric acid, potassium, phosphorus, and calcium, and lactate dehydrogenase
3. Bone marrow aspiration & biopsy: The presence of 30% or more blasts in marrow is s/o acute leukemia
4. Lumbar puncture
5. Cytological & Molecular diagnosis
6. Ultrasonography: testis, kidneys
7. Chest X-ray
Chemotherapy: is the mainstay of treatment
Induction, CNS prophylaxis, consolidation & Maintenance therapy
Surgical role: Central line insertion for chemotherapy
Introduction: Thyroid carcinomas in children represents 1-1.5% of all tumors before the age of 15 years. The incidence of thyroid carcinoma is 2-3 times more in girls than boys & commonly occurs between 7-12 years of age. Radiation to the neck in the childhood is established causative factor in development of thyroid cancer.
Clinical presentation:
1. anterior cervical lymphadenopathy
2. asymptomatic neck mass
3. vocal card palsy (rare)
4. breathlessness or dysphagia because of compression on trachea/ esophagus (rare)
5. Family h/o thyroid cancer (Especially in Medullary cancer)
Main Types:
1. Papillary (most common in children)
2.Follicular
3. Medullary
4. Anaplastic
Physical Examination:
1. Firm painless thyroid nodule in one or both lobes
2. may be associated with lymphadenpathy
3. signs of compression or immobility due to fixation is rare but can be present
4. hoarseness of voice due to vocal cord palsy is again rare in children
Diagnosis:
1. T3, T4, TSH ( Euthyroid)
2. Antithyroid antibodies (to rule out thyroidities)
3. Thyroglobulin levels (for postoperative monitoring)
4. Calcitonin (medullary carcinoma)
5. Ultrasonography of neck
6. Thyroid scan- cold nodules
7. needle biopsy or excision biopsy
8. Chest x-ray/ CT Chest to rule out metastasis
Treatment:
1. Surgery- is the mainstay of treatment
Total thyroidectomy
2. Radioiodine- For the cancer spread outside the thyroid gland
3. Chemotherapy containing low dose Doxorubicin & external irradiation are reserved for anaplastic carcinoma or recurrence of differentiated carcinomas.
Prognosis: Prognosis of differentiated carcinomas is very good & more than 90% survival has been reported.
Sacrococcygeal teratoma is the tumor arising in sacrococcygeal region & it is the commonest tumor found in newborns. It is also seen in infants, children & very rarely in adults. The SCT is more common in girls than boys with ratio of 3:1. The routine use of prenatal ultrasound has made the diagnosis early during fetal life.
Symptoms:
1. Sacral mass
2. Mass in the abdomen & perineum
3. Distension of abdomen
4. Displacement of anus due to sacral mass
5. Constipation
6. Sacral sinus.
Classification: Altaman's classification
Type 1- Entirely outside
Type 2- Mostly outside
Type 3-Mostly inside
Type 4- Entirely inside
Diagnosis:
1. Prenatal Ultrasound- Solid/ cystic mass occupying abdomen as well as perineum
2. CT Scan abdomino-pelvic region/ MRI abdomino-pelvic region
3. Tumor markers- AFP or Alfafetoproteins
Treatment:
1. Surgical excision in benign or mature teratoma
2. Associated with chemotherapy in malignant or immature teratoma
Chemotherapy:
Bleomycin, Etoposide & Cisplatin (BEP) protocol is the commonest first line protocol used.
Prognosis- Good if complete surgical excision is done along with removal of coccyx.
Infantile fibromatosis, originally described by Stout in 1954, seems to be genetic in origin although the mode of transmission is not clear. It refers to a family of soft tissue lesions characterized by proliferation of benign fibrous tissue, which is composed of uniform, elongated, fusiform, or spindle shaped cells surrounded and separated by abundant collagen. According to Enzinger and Weiss, they are classified into superficial and deep fibromatosis. Superficial tumors are usually purplish red as a result of intense vascularity. The intraabdominal organ involvement is also known which carries worse prognosis.
Fibromatosis is a group of related conditions having following common features; proliferation of well differentiated fibroblasts, infiltrative pattern of growth, presence of variable amount of collagen between the proliferating cells, lack of cytological features of malignancy and scanty or absent mitotic activity, aggressive clinical behavior characterized by repeated local recurrences but lack of capacity to metastasize distantly.
In infantile fibromatosis, the Characteristic feature is the presence of small, round intracytoplasmic inclusions. They are periodic acid- Schiff (PAS) negative and they apparently consist of actin filaments7. Morphologically, these lesions occur as unicentric gray white, firm poorly demarcated masses varying from 1 to 15 cm in greatest dimension.
Although histologically benign, this lesion tends to gradually infiltrate in skin subcutaneous tissue, muscles, nerves, blood vessels and even bone. They are rubbery, & tough. Clinically the presentation is of a slow-growing, firm, poorly circumscribed mass. Diagnosis is usually made histologically.
The progression is unpredictable. Wide local excision can provide cure if excision is complete with an adequate resection margin but this may be difficult due to infiltrative nature. Local recurrence occur usually within 18 months of original surgery. Wide local excision is the treatment of choice. Imaging is required for surgical planning. Radiographic examination reveals a soft tissue mass, usually noncalcified. CT appearances are nonspecific; MRI is the best modality for diagnosis & follow-up, specifically in deep fibromatosis.
The primary goal of treatment for fibromatosis is complete surgical resection to achieve negative margins. The management of children with unresectable or recurrent fibromatosis requires a multidisciplinary approach. Nonaggressive therapy with tamoxifen and diclofenac may be the first treatment choice in these patients, but in patients with progressive disease, cytotoxic chemotherapy is indicated. Weekly administration of vinblastine and methotrexate seems to be safe and effective in these children.
Children with cancer require long-term treatment. To administer intravenous drugs of chemotherapy, antibiotics or blood products, pricking every time is not a good option.
For this purpose, there are central lines, which are long tubes, placed through a peripheral vein or central vein into heart. These lines generally stay for longer duration & patient can move around with these catheters.
Types: 1. PICC (peripherally inserted central catheter)
2. Central venous tunneled catheter
3. Implantable ports
PICC- These catheters are inserted through a vein in elbow & advanced into heart. Advantages are they require local anaesthesia, easy to insert, not much expertise required, can be done bedside. Disadvantages are in very small child (less than year) peripheral veins are not big so difficult to put, require proper care time to time to avoid breakage & infection by a trained person.
Central venous tunneled catheter- Centrally inserted tunneled catheters are one of the mostly opted options. It is inserted under anaesthesia through a big vein in the neck or collarbone & the catheter is taken out through a tunneled subcutaneous tract. Advantages are it can be inserted in any age group, can be kept for longer duration until the treatment is over. Disadvantages are require anaesthesia, require aseptic care to avoid tract/ catheter related infection & require expertise to insert.
Implantable ports- Implantable ports are one of the best-suited options for child. There is small port along with a tube. The tube is inserted as any other central catheter & the attached port is placed in the subcutaneous space. Advantages are there is less risk of infection, no disturbances in day-to-day activities & require little care. The disadvantages are require anaesthesia, expertise & repeated needle pricks to access port.
MNTI was first described by Krompecker in 1918 as a congenital melanocarcinoma. It was known by many names as its cellular origin was not clear. These names included pigmented ameloblastoma, retinal anlagetumor, melanotic adamantinoma, retinal choristoma,melanotic progonoma, melanotic epithelial odontoma,pigmented teratoma, atypical melanoblastoma,pigmented epulis and retinoblastic teratoma. Someauthors proved that this tumor causes a high urinaryexcretion of vanillylmandelic acid (VMA), suggestinga neural crest origin. Hence, they coined the term 'melanocytic neuroectodermal tumor of infancy'.
About 200 cases of MNTI have been reported until now. It is found to characteristically occur in the maxilla, more so from the intraoral side. Bone destruction and displacement of teeth often occur because of the intraosseous location in the maxilla. Other sites are the skull, mandible and the brain. The lesion is usually solitary and the mucosa over the lesion is usually intact. It has got a typical bluish color due to the presence of melanin. Although it is a benign tumor with a 2% chance of malignancy, it is locally aggressive. The majority of MNTI patients (there is no sexual predilection) present in the 1 year of life. The children present with swelling in the oral cavity, which often hinders feeding. The differential diagnosis is ameloblastoma, odontoma, odontogenic myxoma, fibroma, rhabdomyosarcoma, EwingÌs sarcoma, LangerhansÌ cell histiocytosis (LCH), non-HodgkinÌs lymphoma.
The plain radiograph of MNTI shows a well-circumscribed radiolucent lesion. As the tumor advances, it destroys the bone suggesting a malignant process. In its typical premaxillary position, the tumor can displace or destroy the developing dentition. CT scanning with intravenous contrast is often used to delineate the margins of osseous involvement. Additionally, MRI can be used to evaluate the bony extent of the lesion. Most MNTIsappear as typical soft tissue tumors with nonenhancing heterogeneous tissue density.
Histopathology shows biphasic pattern with the larger pigmented, melanocyte-like cells and smaller, nonpigmented neuroblast-like cells. Immunohistochemistry (IHC) is positive for cytokeratin, synaptophysin, HMB45, NSE, epithelial membrane antigen, glial fibrillary acidic protein and Leu-7.
The treatment of choice in MNTI is usually complete surgical excision. This treatment can usually be accomplished with a partial maxillectomy by using a Weber-Fergusson incision and a facial degloving approach. The adjacent bone and developing teeth must be sacrificed to get an at least 5 mm margin of healthy tissue. The average local recurrence rate is 15-20%. Radiotherapy and combination chemotherapy including vinblastine, ifosphamide, etoposide, cyclophosphamide, doxorubicin and dactinomycin has been advocated for inoperable recurrence or margin-positive resection. A high index of suspicion is necessary to diagnose this tumor and close follow-up is necessary to detect recurrence. Permanent reconstruction can be done after growth is completed.
Introduction: It is rare benign soft tissue tumor arising from embryonal fat. It is usually seen in infants & children. There are two types, the encapsulated/ well circumscribed is called as lipoblastoma while noncapsulate, diffuse & infiltrating type is called as lipoblastomatosis.
Sites of Origin: The most common site is limb but it can occure in retroperitoneum, head & neck, mediastinum, trunk etc.
Clinical presentation:
1. Painless soft tissue tumor
2. Pressure symptoms
Signs:
1. soft tissue mass
2. pseudofluctuation
3. nontender
4. lobulated surface
Histological subtypes:
1. classic type
2. myxoid lipoblastomas
3. lipoma-like lipoblastomas
4. hibernoma-like lipoblastomas
Treatment: Complete surgical excision is the main modality of treatment. There is no adjuvant therapy is required. The chances of local recurrence is high especially in incomplete excision or when the margins are microscopically involved. So long follow-up is necessary.
Prognosis: Excellent.
Introduction:
Rhabdoid tumor is one of the most aggressive tumor in children. Initially it was considered as an aggressive form (Rhabdomyoblastic variant) of Wilms’ tumor as it arises in kidney but later it is separately classified. When the survival rate of Wilms’ tumor exceeds more than 85%, Rhabdoid tumor survival rate is only 20-25% inspite of all therapies. Apart from kidney, it also occurs in brain, liver, soft tissues, lung, skin, and heart. The median age of occurrence is 11 months & majority cases occur below 3 years of age. Here we will describe only Rhabdoid tumor of kidney.
Clinical Presentation:
1. Haematuria
2. Abdominal distension/ mass
3. Fever
4. Symptoms of brain involvement
Enlarged head
Vomitings
Convulsions
Signs:
1. A palpable mass in abdomen
2. High blood pressure
3. signs of raised intracranial pressure (if associated with brain involvement)
Diagnosis:
1. Complete blood count
2. Urine- routine & microscopy
3. Renal function tests
4. Ultrasound of abdomen
5. USG Doppler
6. CT/MRI abdomen
7. CT/MRI brain
8. CT chest
9. Bone scan
10. CT guided FNAC/ Biopsy of mass
Staging:
Stage I
Tumor is limited to the kidney and completely excised. The renal capsule is intact. The tumor is not ruptured or sampled for biopsy before it is removed. (Fine-needle aspiration is excluded from this restriction.) The vessels of the renal sinus are not involved. No evidence suggests tumor at or beyond the margins of resection.
Stage II
The tumor extended beyond the kidney, but it was completely excised. The tumor may regionally extend into the renal sinus or penetrate the renal capsule. Blood vessels outside the renal sinus may contain tumor, but the tumor must be removed en bloc with the tumor. No evidence of tumor at or beyond the margins of resection is present.
Stage III
Residual nonhematogenous tumor is confined to the abdomen. Any of the following may occur: (1) Tumor involves abdominal lymph nodes. (2) The tumor has penetrated the peritoneal surface. (3) Tumor implants are found on the peritoneal surface. (4) Gross or microscopic tumor remains after surgery. (5) The tumor is not completely resectable because of local infiltration of vital structures. (6) Tumoral spillage occurs before or during surgery. (7) Tumor biopsy was performed before resection.
Stage IV
Hematogenous metastases or lymph node metastases are present outside the abdominal and/or pelvic cavity.
Stage V
Tumors are bilateral.
Management:
1. Chemotherapy: Neoadjuvant chemotherapy (not standardized)
ICE (Ifosphamide + Carboplatin + Etoposide) alternating with VAC
(Vincristine + Adriamycin + Cyclophosphamide)
2. Surgery – Radical nephrectomy with retroperitoneal lymph node
Sampling.
3. Radiotherapy- Flank radiation & lung bath in cases of pulmonary
metastasis
Prognosis:
Despite of all the multimodality treatment, the prognosis remains poor.

Introduction:
Osteosarcoma is one of the common types of bone cancer. Though Osteosarcoma can develop in any part of the body, the most common sites are around the knee joint and close to the shoulder. As it develops from osteoblasts (the cells of developing bone), it commonly occurs in children. It is probably caused by genetic factors which changes immature bone cells into cancer cells.
Osteosarcoma is one of the common types of bone cancer. Though Osteosarcoma can develop in any part of the body, the most common sites are around the knee joint and close to the shoulder. As it develops from osteoblasts (the cells of developing bone), it commonly occurs in children. It is probably caused by genetic factors which changes immature bone cells into cancer cells.
Clinical Presentation:
1. pain and swelling in leg or arm
2. limping gait
3. fracture with trivial trauma
4. backache
5. loss of bladder/ bowel control ( pelvic osteosarcoma)
Signs:
1. swelling just above or below knee joint/ upper arm/ near shoulder
2. painful & limited movements of joint
3. limping gait in case of leg swelling
4. signs of incontinence of bladder/ bowel in case of pelvic or spinal osteosarcoma
5. chest signs in case of metastatic or advanced disease
Diagnosis:
1. X-ray of bone- Lytic lesion with indistinct margins or ossification in the soft tissue with sunburst pattern. Reactive new bone formation is seen as a periosteal elevation, a sign named as a ‘Codman’s angle’ or ‘Codman’s triangle’.
2. MRI of involved bone
3. CT chest
4. Bone scan
5. Tru-cut needle biopsy of tumor
Types
1. Central Osteosarcoma
Conventional
Telangiectatic
Small cell
Low grade
2. Surface Osteosarcoma
Paraosteal
Periosteal
High grade
Treatment:
1. Surgical: Surgical treatments for osteosarcoma consist of either amputation or limb-salvage surgery. Nowadays Limb salvage surgery preferred over amputation. In case of metastatic disease in the lungs, surgery is done to do pulmonary metastatecomy.
2. Chemotherapy: Chemotherapy is given before as well as after surgery. This is called Neoadjuvant chemotherapy and Adjuvant chemotherapy. It removes the microscopic disease in the body.
3. Radiotherapy: The use of radiotherapy is limited in osteosarcoma as it is not very effective. It is used in osteosarcoma that can not be removed surgically.
Prognosis:
In localized disease 60 to 80% survival rate has been reported. Limb osteosarcoma has better prognosis than flat bone osteosarcoma.
1. pain and swelling in leg or arm
2. limping gait
3. fracture with trivial trauma
4. backache
5. loss of bladder/ bowel control ( pelvic osteosarcoma)
Signs:
1. swelling just above or below knee joint/ upper arm/ near shoulder
2. painful & limited movements of joint
3. limping gait in case of leg swelling
4. signs of incontinence of bladder/ bowel in case of pelvic or spinal osteosarcoma
5. chest signs in case of metastatic or advanced disease
Diagnosis:
1. X-ray of bone- Lytic lesion with indistinct margins or ossification in the soft tissue with sunburst pattern. Reactive new bone formation is seen as a periosteal elevation, a sign named as a ‘Codman’s angle’ or ‘Codman’s triangle’.
2. MRI of involved bone
3. CT chest
4. Bone scan
5. Tru-cut needle biopsy of tumor
Types
1. Central Osteosarcoma
Conventional
Telangiectatic
Small cell
Low grade
2. Surface Osteosarcoma
Paraosteal
Periosteal
High grade
Treatment:
1. Surgical: Surgical treatments for osteosarcoma consist of either amputation or limb-salvage surgery. Nowadays Limb salvage surgery preferred over amputation. In case of metastatic disease in the lungs, surgery is done to do pulmonary metastatecomy.
2. Chemotherapy: Chemotherapy is given before as well as after surgery. This is called Neoadjuvant chemotherapy and Adjuvant chemotherapy. It removes the microscopic disease in the body.
3. Radiotherapy: The use of radiotherapy is limited in osteosarcoma as it is not very effective. It is used in osteosarcoma that can not be removed surgically.
Prognosis:
In localized disease 60 to 80% survival rate has been reported. Limb osteosarcoma has better prognosis than flat bone osteosarcoma.

Introduction:
Ovarian Carcinomas are unusual in pediatric age group. It accounts for only 1% of all tumors in girls below the age of 17 years. In the first differential diagnosis of ovarian tumors in adolescent girls, germ cell tumor comes first and carcinomas comes last.
Ovarian Carcinomas are unusual in pediatric age group. It accounts for only 1% of all tumors in girls below the age of 17 years. In the first differential diagnosis of ovarian tumors in adolescent girls, germ cell tumor comes first and carcinomas comes last.
Clinical Presentation:
Abdominal distension
Lower abdominal pain
Weight loss
Malaise
Signs:
Grossly underweight
Lower abdominal mass
ascites
Diagnosis:
Tumor markers- CA 125
CA 125 - CA 125 serology has only 78.1 % sensitivity & 76.8 % specificity but it is useful in identifying recurrent or residual disease.
Ultrasonography- solid, cystic or mixed; arising from, areas of hemorrhage, liver metastasis, retroperitoneal lymph nodes.
CT scan- extent of tumor, exact origin of tumor, better delineation, lymph node status, peritoneal metastasis
Bone scan-for detection of metastasis.
Histopathology:
Serous
Mucinous
Undifferentiated
Treatment:
Surgery: is done to confirm the diagnosis, stage the disease, & achieve tumor clearance in early stages & cytoreduction in late cases.
Chemotherapy: Cisplatin, cyclophosphamide, carboplatin, bleomycin, etoposide, & paclitaxel in various combinations.
Prognosis:
Stage of the disease
Histology
However the prognosis in premenarchal girls appears to be poor with no reported long term survivors.
Abdominal distension
Lower abdominal pain
Weight loss
Malaise
Signs:
Grossly underweight
Lower abdominal mass
ascites
Diagnosis:
Tumor markers- CA 125
CA 125 - CA 125 serology has only 78.1 % sensitivity & 76.8 % specificity but it is useful in identifying recurrent or residual disease.
Ultrasonography- solid, cystic or mixed; arising from, areas of hemorrhage, liver metastasis, retroperitoneal lymph nodes.
CT scan- extent of tumor, exact origin of tumor, better delineation, lymph node status, peritoneal metastasis
Bone scan-for detection of metastasis.
Histopathology:
Serous
Mucinous
Undifferentiated
Treatment:
Surgery: is done to confirm the diagnosis, stage the disease, & achieve tumor clearance in early stages & cytoreduction in late cases.
Chemotherapy: Cisplatin, cyclophosphamide, carboplatin, bleomycin, etoposide, & paclitaxel in various combinations.
Prognosis:
Stage of the disease
Histology
However the prognosis in premenarchal girls appears to be poor with no reported long term survivors.

Introduction:
Germ Cell Tumors are the neoplasm arising from primordial germ cells which produces specialized cells in the body like sperm and egg cells. With the recent advances in cisplatin based chemotherapy, the cure rate of germ cell tumors is increased if diagnosed in early stages. It frequently occurs in three modal peaks of life, infancy, 25-40 yrs and around 60 yrs.
Germ Cell Tumors are the neoplasm arising from primordial germ cells which produces specialized cells in the body like sperm and egg cells. With the recent advances in cisplatin based chemotherapy, the cure rate of germ cell tumors is increased if diagnosed in early stages. It frequently occurs in three modal peaks of life, infancy, 25-40 yrs and around 60 yrs.
Sites:
Testes
Ovaries
Paratesticular area
Abdomen (retroperitoneum)
Mediastinum
Brain
Types:Malignant GCTs: The tumors such as yolk sac tumors, choriocarcinoma, and immature tearatomas encompass this type. The elevated tumor markers, rapid growth signifies malignant transformation.
Benign GCTs: Teratomas are benign tumors. They have characteristic appearance where there are teeth, bone, hair is found inside the tumor.
Symptoms:
Painless scrotal mass
Abdominal mass
Abdominal pain
Breathlessness
Sacral mass
Investigations:
Tumor markers like AFP, β-HCG, LDH
Biopsy
CT Scan/ MRI
Treatment:Surgery: The location of the tumor may influence the need for surgery. When possible, the first choice is usually to try and remove the entire tumor. This can be enough to cure most teratomas and immature teratomas. The coccyx needs to be removed in case of sacrococcygeal teratomas.
Chemotherapy: BEP (Bleomycin, Etoposide & Cisplatin) is the first line chemotherapy for the malignant germ cell tumors. This tumors are very much chemosensitive & the fall in tumor marker gives the idea about the response. If the complete resection is not possible initially then neoadjuvant chemotherapy is advisable.
Radiotherapy: It is indicated as a local therapy when the surgery is not possible.
Prognosis:
The 5-year survival rate is about 95% in germ cell tumors.
Testes
Ovaries
Paratesticular area
Abdomen (retroperitoneum)
Mediastinum
Brain
Types:Malignant GCTs: The tumors such as yolk sac tumors, choriocarcinoma, and immature tearatomas encompass this type. The elevated tumor markers, rapid growth signifies malignant transformation.
Benign GCTs: Teratomas are benign tumors. They have characteristic appearance where there are teeth, bone, hair is found inside the tumor.
Symptoms:
Painless scrotal mass
Abdominal mass
Abdominal pain
Breathlessness
Sacral mass
Investigations:
Tumor markers like AFP, β-HCG, LDH
Biopsy
CT Scan/ MRI
Treatment:Surgery: The location of the tumor may influence the need for surgery. When possible, the first choice is usually to try and remove the entire tumor. This can be enough to cure most teratomas and immature teratomas. The coccyx needs to be removed in case of sacrococcygeal teratomas.
Chemotherapy: BEP (Bleomycin, Etoposide & Cisplatin) is the first line chemotherapy for the malignant germ cell tumors. This tumors are very much chemosensitive & the fall in tumor marker gives the idea about the response. If the complete resection is not possible initially then neoadjuvant chemotherapy is advisable.
Radiotherapy: It is indicated as a local therapy when the surgery is not possible.
Prognosis:
The 5-year survival rate is about 95% in germ cell tumors.

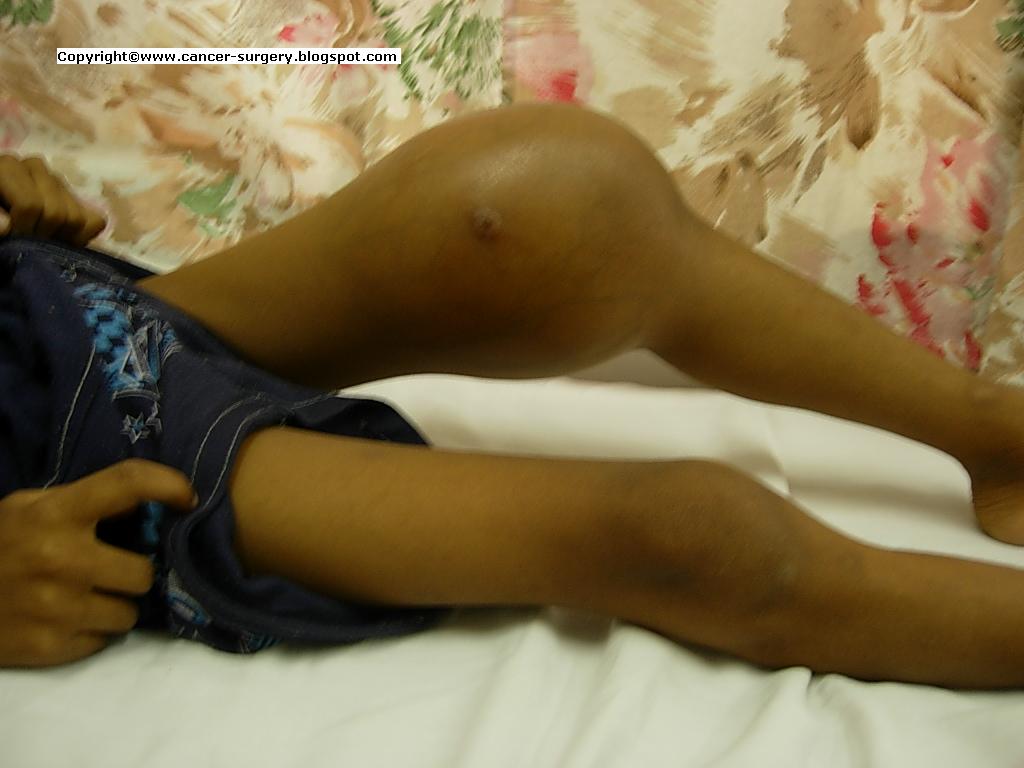
Introduction:
Rhabdomyosarcoma also called in short as RMS is a malignancy of mesenchymal origin. It is a tumor of muscle cells called rhabdomyoblasts. It is third most common solid tumor of childhood.
Incidence:
In the United States, about 350 new cases are diagnosed each year in children. Almost two-thirds of RMS cases develop in children under the age of 10. It accounts for more than half of soft tissue sarcomas of childhood.
Associated conditions & predisposing factors:
· Li-Fraumeni syndrome
· Neurofibromatosis type 1
· Beckwith-Wiedemann syndrome
· Cardio-facio-cutaneous syndrome
· Costello syndrome
· Maternal & paternal abuse of cocaine and marijuana
Sites:
Parameningeal- Adjacent to the base of the skull
Orbital - Around the eye
Head and neck
Arms and legs (extremities)
Urinary system and reproductive organs (bladder, vagina, prostate and paratestes sites)
Types of RMS:
Embryonal
Alveolar
Pleomorphic
Undifferentiated
Symptoms:
Rapidly enlarging firm mass
Haematuria,
Dysuria,
Vaginal mass or bleeding
Scrotal mass
Periorbital swelling
Nasal discharge
Lump in abdomen
Investigations:
CT scan or MRI of local site
Bone marrow studies
CT scan chest
Bone scan
Biopsy of the tumor
PET scan
Histopathology: Rhabdomyosarcoma is a small round blue cell tumor
Treatment:
1. Surgery: If the tumor can be completely resectable with achievement of negative margins, then surgery should be done upfront.
2. Chemotherapy: It is used as neo-adjuvant or adjuvant setting. The drugs used are
Vincristine
Ifosphamide
Etoposide
Adriamycin
Cyclophosphamide
Dactinomycin
3. Radiotherapy: It is used to minimize local recurrence in high risk cases.
Prognostic factors:
5-Year survival of RMS is increased over 70%.
Prognostic factors include
Extent of the disease
Primary tumor site: favorable sites are the orbit, head and neck and bladder, all other sites including parameningeal tumors and non-bladder genitourinary sites are classified unfavorable.
Histology subtype: Embryonal & pleomorphic histology tumors have a more favorable outcome while alveolar & undifferentiated has unfavorable outcome.
Age: Patients under one and over 10 years of age have a somewhat less favorable prognosis.
Presence of Distant Metastasis: Metastatic at presentation has a less favorable prognosis.

Introduction:
It is the second most common solid tumor in children after brain tumor. It occurs in the neural crest cells, called neuroblasts, of the sympathetic nervous system. It can occur in a number of anatomical sites. The majority of tumors (65%) are located in the abdomen, often in one of the two adrenal glands. However, primary tumors can occur anywhere in the body. Other common sites are the chest, neck or pelvis. It is one of the most malignant neoplasms of children & rarely seen in adults.
It is the second most common solid tumor in children after brain tumor. It occurs in the neural crest cells, called neuroblasts, of the sympathetic nervous system. It can occur in a number of anatomical sites. The majority of tumors (65%) are located in the abdomen, often in one of the two adrenal glands. However, primary tumors can occur anywhere in the body. Other common sites are the chest, neck or pelvis. It is one of the most malignant neoplasms of children & rarely seen in adults.
Incidence:
Neuroblastoma represents 7.5% of all childhood cancers. It is most commonly diagnosed between 17 months and 2 years of age.
Aetiology:
Neuroblastoma develops when normal neuroblasts (the immature cells of the sympathetic nervous system) fail to mature into nerve cells. Instead, they continue to grow leading to mass of cancer cells.
Other hypothesis is “mutations” occurring in Neuroblastoma cells.
Symptoms:
Symptoms of Neuroblastoma are dependent upon the site, metastatic disease or associated paraneoplastic syndromes,
Abdominal distention/constipation
Pain in the abdomen.
Fever, bony pain, anemia, if the disease has spread to the bone marrow, causing a decrease in red blood cells.
Bulging, darkening, proptosis or swelling of the eye, if the disease has spread to tissues and bone around the eyes.
Persistent diarrhea, high blood pressure, palpitations, reddening of the skin, sweating, involuntary movements of eyes & limbs, if there is associated paraneoplastic syndrome
Neuroblastoma represents 7.5% of all childhood cancers. It is most commonly diagnosed between 17 months and 2 years of age.
Aetiology:
Neuroblastoma develops when normal neuroblasts (the immature cells of the sympathetic nervous system) fail to mature into nerve cells. Instead, they continue to grow leading to mass of cancer cells.
Other hypothesis is “mutations” occurring in Neuroblastoma cells.
Symptoms:
Symptoms of Neuroblastoma are dependent upon the site, metastatic disease or associated paraneoplastic syndromes,
Abdominal distention/constipation
Pain in the abdomen.
Fever, bony pain, anemia, if the disease has spread to the bone marrow, causing a decrease in red blood cells.
Bulging, darkening, proptosis or swelling of the eye, if the disease has spread to tissues and bone around the eyes.
Persistent diarrhea, high blood pressure, palpitations, reddening of the skin, sweating, involuntary movements of eyes & limbs, if there is associated paraneoplastic syndrome
Investigations:
Complete blood count
Biochemistry including serum LDH & serum ferritin
Bone marrow aspiration & Biopsy
Urinary VMA (Vanillyl mandelic acid) and HMV (Homovanillic acid)
N-myc amplification
DNA index
MIBG (131I-meta-iodobenzyl guanidine) scan
Chest X-Ray
CT Scan of primary region
MRI spine ( If neurological symptoms)
Bone scan
Biopsy of the tumor
Prognostic factors:
Age of the child
Stage of the disease
Tumor Histopathology
Myc-N Status
DNA Index (Ploidy)
Treatment:
Induction Chemotherapy: Multi-drug chemotherapy is first given to try to reduce the size of the primary and metastatic tumor(s). The drugs used are cyclophosphamide, doxorubicin, cisplatin, etoposide, Ifosphamide.
Surgery: Surgery is done to remove as much tumor as safely possible. In some locations its possible to remove whole mass
Radiation Therapy: It is usually given locally to the tumor site.
Myleoablative therapy followed by stem cell or bone marrow transplant: Intensive doses of chemotherapy to eliminate any remaining tumor cells, followed by an autologous peripheral blood stem cell transplant, or sometimes a bone marrow transplant, to restore the immune system is used in high risk disease.
Minimum residual disease therapy: Additional drugs like 13-cis-Retinoic acid, aimed at eliminating or altering minimal residual disease is given after consolidation therapy.
MIBG Therapy: It is used when surgery is not possible because of persistent bone marrow disease after induction chemotherapy.
Prognosis:
Low risk patients have a greater than 90% survival.
Intermediate risk patients have a 70-90% chance of survival.
High risk patients have a 30% chance of survival
Intermediate risk patients have a 70-90% chance of survival.
High risk patients have a 30% chance of survival

Introduction:
Bone tumors are sixth most common group of cancers in children. Two common types of primary bone cancers found in children are osteosarcoma & Ewing sarcoma. Ewing sarcoma is the second most common tumor of the bone. It most often affects bones of the pelvis, the tibia, fibula, and femur, and can also begin in the soft tissues. This disease most often occurs in second sdecade of life that is between the ages of 10 and 20.They are small round cell undifferentiated tumors.
Bone tumors are sixth most common group of cancers in children. Two common types of primary bone cancers found in children are osteosarcoma & Ewing sarcoma. Ewing sarcoma is the second most common tumor of the bone. It most often affects bones of the pelvis, the tibia, fibula, and femur, and can also begin in the soft tissues. This disease most often occurs in second sdecade of life that is between the ages of 10 and 20.They are small round cell undifferentiated tumors.
Types:
These are the Ewing Family of tumors which include Ewing sarcoma and PNET (Primitive neuroectodermal Tumors.
Symptoms:
1. Pain at the site of tumor
2. Swelling at the site of tumor
3. Weight loss
4. Anorexia
5. fever, malaise
6. cough
Prognostic factors:
1. Site of the tumor
2. The presence of metastasis
3. Size of the tumor
4. Age
5. Erythrocyte Sedimentation rate
6. Serum LDH levels
Investigations:
1. Plain X-Ray of the local site
2. CT scan or MRI of the primary tumor
3. Chest X-Ray
4. CT of the chest
5. Bone scan
6. Bone marrow aspiration and bone marrow biopsy
Treatment:
Ewing sarcoma is treated with multimodality treatment.
1. Surgery:
The options are
Amputation
Rotationplasty
Limb salvage surgery
Surgery is not effective for treating metastatic disease.
2. Chemotherapy:
Chemotherapy uses drugs like Ifosfamide, Etoposide, Adriamycin, Vincristine, Cyclophosphamide.It may be used before surgery to shrink a tumor so that it can be removed surgically, and will be used after surgery for any remaining cancer cells.
3. Radiation therapy:
Ewing sarcoma is very sensitive to radiation.When surgery is not possible because of large tumor mass, metastatic disease, certain site of cancer then radiation therapy is an effective answer.
These are the Ewing Family of tumors which include Ewing sarcoma and PNET (Primitive neuroectodermal Tumors.
Symptoms:
1. Pain at the site of tumor
2. Swelling at the site of tumor
3. Weight loss
4. Anorexia
5. fever, malaise
6. cough
Prognostic factors:
1. Site of the tumor
2. The presence of metastasis
3. Size of the tumor
4. Age
5. Erythrocyte Sedimentation rate
6. Serum LDH levels
Investigations:
1. Plain X-Ray of the local site
2. CT scan or MRI of the primary tumor
3. Chest X-Ray
4. CT of the chest
5. Bone scan
6. Bone marrow aspiration and bone marrow biopsy
Treatment:
Ewing sarcoma is treated with multimodality treatment.
1. Surgery:
The options are
Amputation
Rotationplasty
Limb salvage surgery
Surgery is not effective for treating metastatic disease.
2. Chemotherapy:
Chemotherapy uses drugs like Ifosfamide, Etoposide, Adriamycin, Vincristine, Cyclophosphamide.It may be used before surgery to shrink a tumor so that it can be removed surgically, and will be used after surgery for any remaining cancer cells.
3. Radiation therapy:
Ewing sarcoma is very sensitive to radiation.When surgery is not possible because of large tumor mass, metastatic disease, certain site of cancer then radiation therapy is an effective answer.
Introduction
This is the most common primary tumor of the eye in children. It arises from the retina. .The retina is a layer of nerve tissue that coats the back of the eye, which is important for vision of person. Retinoblasts (immature cells of the retina) multiply during gestation and early life, to make enough cells to create the retina. As child grows, these cells mature & no longer differentiate. If these immature retinoblasts turn into cancer cells, retinoblastoma develops, the cause of which is unknown. The gene responsible is RB1 gene.
Incidence:
The annual incidence is one in 20000 children. It occurs most often in children under 4 years of age.
Types:
Hereditary:
•Hereditary form of Retinoblastoma occurs in 40%.
•May have more than one tumor
•Tumor often affects both eyes
•May have tumors in other parts of the body
•At increased risk for other cancers later in life
Non-hereditary:
•Most children with retinoblastoma (60%) do not have the genetic form.
•They develop tumor in only one eye i.e. unilateral.
•These children do not have an increased risk of developing other cancers.
•Their offspring have the same risk of developing retinoblastoma as other children in the population.
Symptoms:
•A pupil that looks white or red instead of the usual black i.e. called as white or cat’s eye reflex.
•A crossed eye i.e. strabismus.
•Poor vision
•A red, painful eye
•An enlarged pupil
•Differently colored irises
Investigations:
•Examination under general anesthesia using Retcam
•Ultrasound
•CT (CAT) scan
•MRI
•CSF examination
•Bone marrow studies
•Bone scan in advanced disease
•Chromosomal analysis (in certain cases)
Management:
1.Surgery to remove the eye, known as enucleation. This s done when there is no vision in eye to save further spread & life. Enucleation may also be recommended if the tumor does not respond to treatment.
2.Chemotherapy is used to shrink tumors in the eye. This approach is often used in children with bilateral disease (both eyes) for saving at least one eye which has less disease. It is also used in small tumors where the eye vision is present to save the eye. in combination with other measures such as
1.Photocoagulation--using laser light to destroy blood vessels supplying the tumor.
2.Thermotherapy--using heat to destroy tumor cells
3.Cryotherapy--using extreme cold to destroy tumor cells
4.Radiation Therapy—It is used for control of local disease with preservation of vision. Radiation plaque therapy and particle beam radiotherapy are used frequently.
Prognosis:
The five-year survival rate for children with retinoblastoma is more than 90%.
Introduction:
Brain tumors are second most common childhood malignancy after leukemia. It contributes to 20% of pediatric cancers. With the improvement in imaging, surgical modalities, radiation therapy & chemotherapy, the outlook of children with brain tumors has improved. These combined modalities give the maximum benefit to the patients however the early diagnosis is important for good results.
Aetiology:
Though it is unclear, certain familial syndromes are associated with brain tumors showing genetic predisposition.
Neurofibromatosis
Ataxia telengiectasia
Tuberous sclerosis
Von Hipple- Lindau disease
Types:
Astrocytoma
Medulloblastoma
Ependymoma
Glioblastoma
Craniopharyngioma
Pineal gland tumors
Choroids plexus tumors
Meningioma
Neuroblastoma
Primitive neuroectodermal tumors
Rhabdoid tumors
Symptoms:
Projectile vomiting
Headache
Irritability
Unable to balance while walking
Weakness of limbs
Convulsions
Behavioral changes
Investigations:
CT/MRI –brain ( Plain & Contrast films)
CSF examination for malignant cells
Complete blood counts
Biochemistry
Chest X-Ray
Treatment:
Surgery-
Complete excision or debulking surgery should be the aim. The ventriculoperitoneal shunt (VP shunt) can be done in sick child for rapid decompression in raised intracranial tension. If the complete excision is not possible stereotactic biopsy or craniotomy with biopsy of the tumor is done.
Radiotherapy-
With the modern radiotherapy (use of linear accelerator) the radiotherapy related complications are minimized. The Radiotherapy should be avoided below 3 years as the brain is still developing. In case of spinal metastasis the RT is also given to the spine.
Chemotherapy- The use of platinum based chemotherapy after surgery and radiotherapy for minimum residual disease improved the prognosis in children with brain tumors.

Introduction:
There are variety of tumors arising from kidney in children but the most common is Wilms' tumor named after Karl Max Wilms who is the first person to review the literature & described the unique properties/ So it is named after him. It is also called as nephroblastoma. It occurs in children upto 6 years with peak at 3 years.It is extremely uncommon after 6 yrs.
There are variety of tumors arising from kidney in children but the most common is Wilms' tumor named after Karl Max Wilms who is the first person to review the literature & described the unique properties/ So it is named after him. It is also called as nephroblastoma. It occurs in children upto 6 years with peak at 3 years.It is extremely uncommon after 6 yrs.
Syptoms:
1. mass or Lump or distension of abdomen ( most common)
2. abdominal pain.
3. hematuria (blood in the urine or smoky urine)
4. fever
5. refusal of feeds
6. lethargy.
7. weight loss
Investigations:
The following investigations are proposed for whole work up when one suspect the Wilms tumor.
1. CT scan of abdomen & CT guided FNAC of tumor
2. X-ray chest
3. Comlete Blood Counts
4. Biochem profile
Treatment:
Treatment for wilms' tumor is multidisciplinary
It includes surgery, chemotherapy & radiotherapy
Surgery- includes radical nephrectomy with lymph node sampling
Chemotherapy- combination of drugs like Vincristine / Actinomycin- D/ Cyclophosphamide.
Radiotherapy- is given to the flank in advanced disease or to the chest if there are metastasis.
Prognosis:This is one of the most curable cancer. the cure rate is more than 80% with multimodality treatment.
Other tumors of kidney in childhood
1. Clear cell sarcoma
2. Rhabdoid tumor
3. Renal cell carcinoma
4. Primitive neuroectodermal tumor
5. Infantile osteoid producing kidney tumor.
1. mass or Lump or distension of abdomen ( most common)
2. abdominal pain.
3. hematuria (blood in the urine or smoky urine)
4. fever
5. refusal of feeds
6. lethargy.
7. weight loss
Investigations:
The following investigations are proposed for whole work up when one suspect the Wilms tumor.
1. CT scan of abdomen & CT guided FNAC of tumor
2. X-ray chest
3. Comlete Blood Counts
4. Biochem profile
Treatment:
Treatment for wilms' tumor is multidisciplinary
It includes surgery, chemotherapy & radiotherapy
Surgery- includes radical nephrectomy with lymph node sampling
Chemotherapy- combination of drugs like Vincristine / Actinomycin- D/ Cyclophosphamide.
Radiotherapy- is given to the flank in advanced disease or to the chest if there are metastasis.
Prognosis:This is one of the most curable cancer. the cure rate is more than 80% with multimodality treatment.
Other tumors of kidney in childhood
1. Clear cell sarcoma
2. Rhabdoid tumor
3. Renal cell carcinoma
4. Primitive neuroectodermal tumor
5. Infantile osteoid producing kidney tumor.

Introduction:
Hodgkin’s disease is a cancer of the lymphatic system. It is also called as Hodgkin’s lymphoma. Hodgkin’s disease accounts for 5% of cancers diagnosed in children. It is rare before the age of five years. The number of cases increases significantly in the second decade of life.
Hodgkin’s disease is a cancer of the lymphatic system. It is also called as Hodgkin’s lymphoma. Hodgkin’s disease accounts for 5% of cancers diagnosed in children. It is rare before the age of five years. The number of cases increases significantly in the second decade of life.
Symptoms:
1. enlarged lymph nodes, called lymphadenopathy
(Painless, firm, rubbery, and movable)
2. loss of appetite
3. loss of weight
4. fever
5. lethargy
6. lump in abdomen
7. pain in abdomen
8. itching
9. night sweat
10. cough or breathlessness.
Investigations:
1. X-ray Chest
2. Ultrasonography / CT scan of abdomen
3. lymph node biopsy
4. Complete Blood Count (specially ESR)
5. Biochemistry ( specific- LDH, β2 macroglobulin, serum albumin)
6. Bone marrow / Bone scan in advanced stage
7. PET scan
Treatment:
Hodgkin’s disease is treated with chemotherapy & radiotherapy. The surgery is done only for the diagnosis.
Chemotherapy:
The following drugs are used
1. Adriamycin
2. Bleomycin
3. Vinblastine
4. Dacarbazine
Radiotherapy:
Radiotherapy is used after 4 or 6 cycles of chemotherapy.
Prognosis: Excellent. 5 year survival rate is >85%.
1. enlarged lymph nodes, called lymphadenopathy
(Painless, firm, rubbery, and movable)
2. loss of appetite
3. loss of weight
4. fever
5. lethargy
6. lump in abdomen
7. pain in abdomen
8. itching
9. night sweat
10. cough or breathlessness.
Investigations:
1. X-ray Chest
2. Ultrasonography / CT scan of abdomen
3. lymph node biopsy
4. Complete Blood Count (specially ESR)
5. Biochemistry ( specific- LDH, β2 macroglobulin, serum albumin)
6. Bone marrow / Bone scan in advanced stage
7. PET scan
Treatment:
Hodgkin’s disease is treated with chemotherapy & radiotherapy. The surgery is done only for the diagnosis.
Chemotherapy:
The following drugs are used
1. Adriamycin
2. Bleomycin
3. Vinblastine
4. Dacarbazine
Radiotherapy:
Radiotherapy is used after 4 or 6 cycles of chemotherapy.
Prognosis: Excellent. 5 year survival rate is >85%.

Introduction:
The liver is a large, very important organ situated in the right upper quadrant of abdomen. The normal functions of the liver in the body are:
To help store nutrients from food
To break down and remove harmful chemicals from the body
To build chemicals that the body needs to stay healthy
Hepatoblastoma: The most common type of liver cancer in children is Hepatoblastoma. it occurs most frequently in infants or very young children between 2 months and 2 year.
Symptoms:
- Lump in the abdomen (most common)
- Reduced appetite
- lethargy
- nausia / vomiting
- Pain in abdomen
- Jaundice
Investigations:
The following minima investigations are proposed to diagnose hepatoblastoma
- Tumor markers- serum AFP
- CT scan of abdomen with Ct guided FNAC or Biopsy
- X-ray chest
- Complete blood count
- Biochem profile
Treatment:
Surgery & chemotherapy are two tratment options
Surgery: Liver resection according to the location & extent of the tumor. It will be either
- Rt hepatectomy
- Lt hepatectomy
- Extended Rt or Lt hepatectomy
- Bi or trisegmentectomy
If liver resection is not possible because of multicentric disease then liver transplant is the only treatment of option.
Chemotherapy:
Chemotherapy is given before or after surgery. Surgery is done either as a sandwitch surgery (in between cycles of chemotherapy) or it is done at the start & then chemotherapy is given. Antitumor drugs dispersed in a lipid lymphographic medium are intraarterially administered unresectable hepatoblastoma.The more information about intra-arterial chemotherapy can be read in Rare Adult & Pediatric Cancer Web Blog
The drugs used are
- Cisplatin
- Doxorubicin
- Vincristine
- 5-Fluorouracil
Other less common malignant liver tumor in children:
Hepatocellular carcinoma.
Subscribe to:
Comments (Atom)








{kind=link}
{kind=link}